BIOINFORMATICS101
Analyzing Gene Ontology data
Gene ontology (GO) is a controlled vocabulary that connects each gene to one or more functions/gene products. Through a GO association file, each gene of a given species of organisms is connected to one or more GO definition files.
The authoritative source for GO definition files is http://geneontology.org
GO definition files can be downloaded on a Linux machine in the following way.
wget http://purl.obolibrary.org/obo/go.obo The above go.obo file has GO definition records of the following form .

Now we can download the GO association file for humans which annotates or maps the human genes to their corresponding GO definition file ids in the following way.
wget http://geneontology.org/gene-associations/goa_human.gaf.gz Now we can uncompress the GO association file:
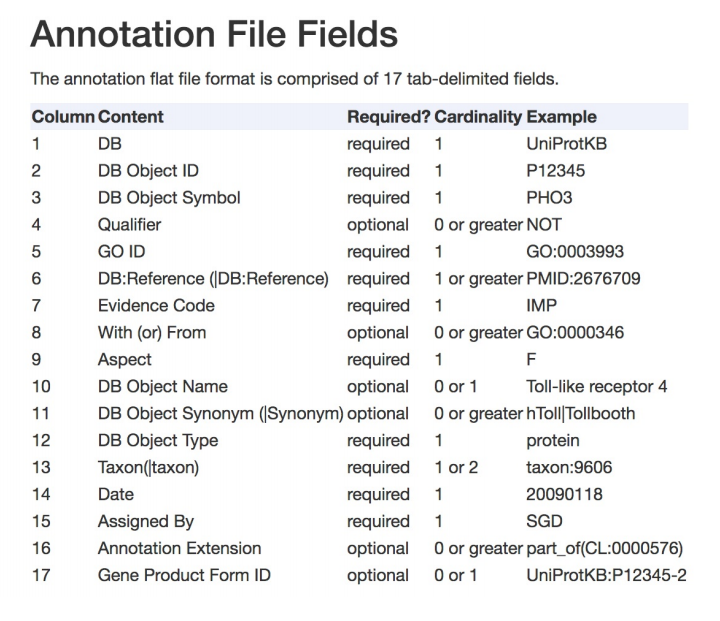
gunzip goa_human.gaf.gz The resulting .gaf file has a flat file format with 17 tab-delimited fields:
Inside the uncompressed goa_human.gaf file, comments start with the ‘!’ character. We can remove lines containing those comments and rewrite the contents of the .gaf file to a new file assoc.txt
cat goa_human.gaf | grep -v '!' > assoc.txt Now we can answer some useful questions simply using the linux commands
- How many gene to product/function mappings are there in the association file?

In the above command we pipe the content of the assoc.txt file to the wc command. With the -l option specified, the wc command finds the number of lines in the assoc.txt file which equals the total number of mappings/associations, which at this time equals 469828. This number can change with time.
- Gene names are in column 3 of the .gaf file. List the first 10 names.
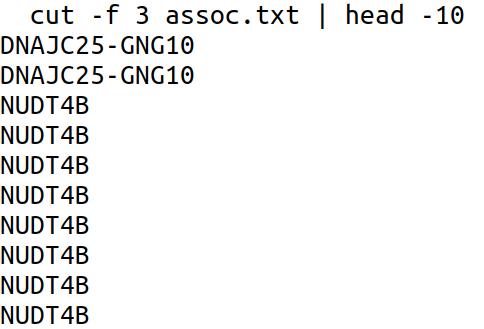
Here we have used the cut command to cut the column 3 (field 3) in the assoc.txt file which is piped to the head command with the option -10 which lists the first 10 lines.
Here we can see that the column #3 has a lot of redundant gene names which takes us to the next question.
- Find the number of unique gene names.
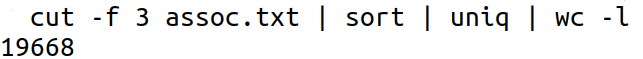
We have piped the content of the third column (i.e. gene names) to the sort command which does a string sort and then pipe the output to the uniq command which removes redundant entries. Then we pipe that output to the wc command with the -l option which counts the number of lines as 19668.
- What are the most highly annotated gene names in the GO dataset?
It would be easier to imagine how this can be achieved if I break it into two steps. First we can find the number of annotations for each gene which can be achieved by:
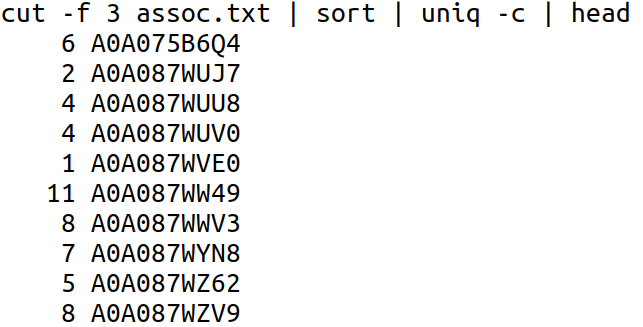
Now we can sort this output by its first column (k1), as numbers (n), from highest to lowest (r) and list the first 20 lines, to find the 20 human gene names with the highest number of gene name to gene product/function mappings.
So now, if I write the whole set of commands in one line to achieve our target:
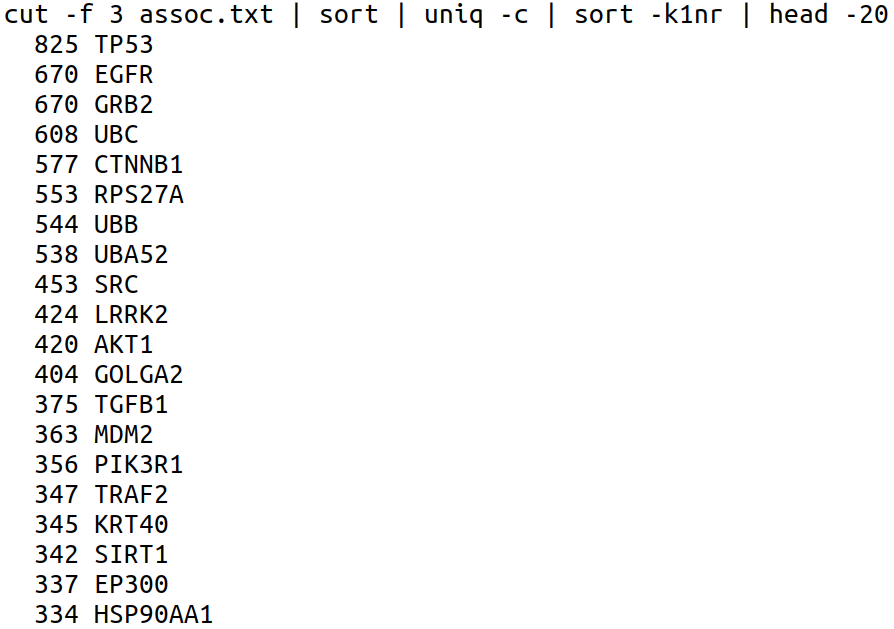
Now we can see that the human tumor suppressor gene TP53 has the highest number of annotations.
- What is the average number of annotations per gene?
First, we can save the output at the end of question #4 into a separate file.
At the end of question #4 we can see that inside the gene_count.txt file there is empty space in front of the first column, and in between the column 1 and column 2, there is a smaller space.
Using:
we can reduce all the whitespaces with variable size to a single whitespace.
Now we can install a very handy tool called datamash for on-the-go simple data analysis by:
Now we can use the output of the tr -s ‘ ‘ command on gene_counts.txt as an input for the datamash command and use the single whitespace that we created as the delimiter to separate the columns. Since there is still a single whitespace in front of the column1 (annotations per gene), column 1 will be interpreted by datamash as column2. Now we can use the mean option in datamash to achieve our target of finding the average annotations per gene.


Like!! I blog frequently and I really thank you for your content. The article has truly peaked my interest.